IN A NUTSHELL
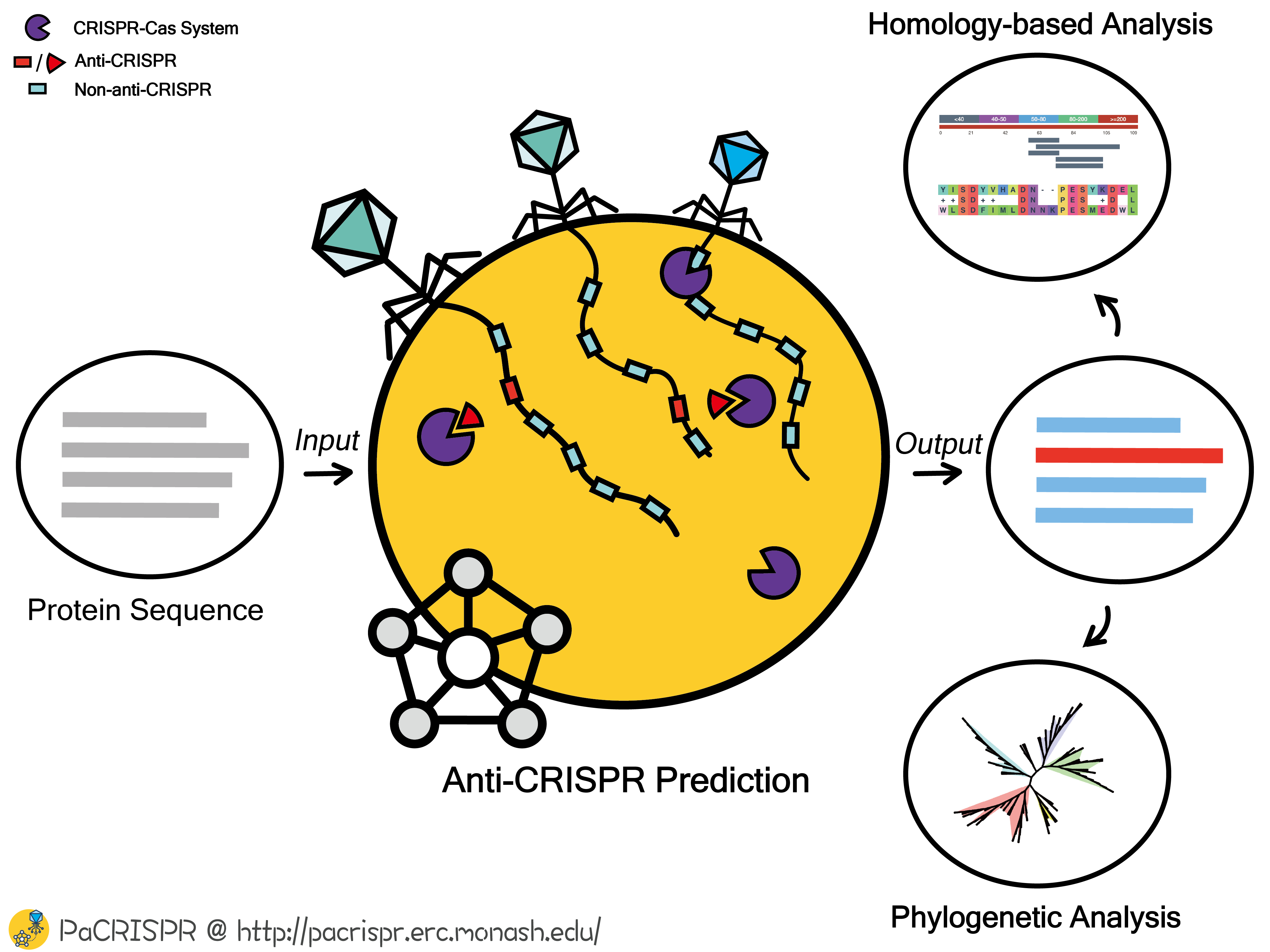
The application of the PaCRISPR server to predict and visualize anti-CRISPR proteins
Anti-CRISPRs are widespread amongst bacteriophage and certain mobile genetic elements (such as transposons and prophage) and by inactivating the bacterial host's CRISPR-Cas defence system, anti-CRISPRs promote bacteriophage infection and horizontal gene transfer. Identifying anti-CRISPR proteins and characterizing specific aspects of their function opens an avenue to explore and control CRISPR-Cas machineries, and therefore facilitate the development of new CRISPR-Cas based biotechnological and therapeutic tools. Past studies have uncovered important anti-CRISPRs in several model phage genomes, but a challenge exists to comprehensively screen and characterize anti-CRISPRs accurately and efficiently from the vastly increasing number of phage genome and metagenome sequencing projects.
In this work, we have developed an ensemble learning based predictor, PaCRISPR, to accurately identify anti-CRISPRs from protein datasets derived from genome and metagenome sequencing projects. PaCRISPR employs different types of feature recognition united within an ensemble framework. Extensive cross-validation and independent tests show that PaCRISPR achieves a significantly more accurate performance compared with homology-based baseline predictors and an existing toolkit. The performance of PaCRISPR was further validated in discovering anti-CRISPRs that were not part of the training for PaCRISPR, but which were recently demonstrated to function as anti-CRISPRs for phage infections within species of Listeria, Enterococcus, Streptococcus, Staphylococcus and Sulfolobus. For each predicted anti-CRISPR, data visualization on its relationships with other anti-CRISPRs, highlighting sequence similarity and phylogenetic considerations, is part of the output from PaCRISPR. In this way, the PaCRISPR toolkit is expected to serve in preliminary screening of potential anti-CRISPRs for experimental validation and functional characterisation.
ARCHITECTURE
The overall workflow of PaCRISPR is presented in terms of data collection and curation, feature encoding, model training and integration, model performance evaluation, and toolkit development and usage.
1. Dataset
- Training dataset: 98 positive and 902 negative samples.
- Independent dataset: 26 positive and 260 negative samples.
- Two pure long non-anti-CRISPR datasets: 266 non-anti-CRISPRs from phages, and 597 non-anti-CRISPRs from bacterial known and putative MGEs.
- Case study: 5 very recently discovered anti-CRISPRs, and a bacterial contig (NCBI-RefSeq: NZ_ALTM01000002.1) from the genome sequence of Streptococcus agalactiae strain GB00548.
2. Feature encoding
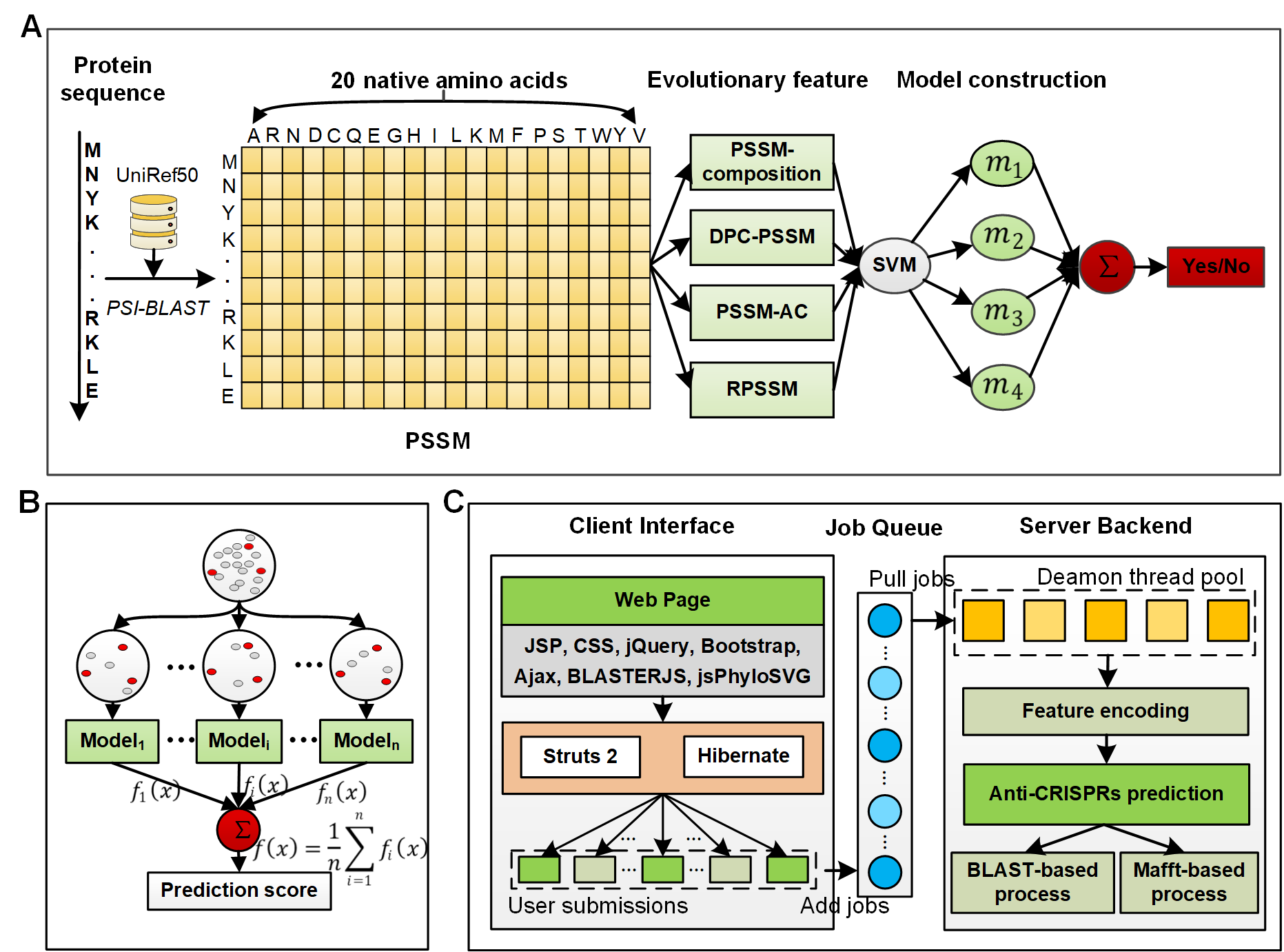
Novel anti-CRISPRs are especially difficult to identify given that they are significantly diverse, sharing no conserved sequence or structural motifs. The low sequence similarity therefore makes it particularly challenging to predict anti-CRISPRs from sequence-based features, which only mine characteristics from protein sequences. Instead, so-called evolutionary features are extracted from the Position-Specific Scoring Matrix (PSSM). To some extent these features track the evolutionary history of proteins and are proposed, therefore, to learn more informative patterns. Evolutionary features have been widely applied and demonstrated to have a significant contribution to protein attribute and function predictions, especially to identify those highly evolved proteins without observed signals.
Using the POSSUM toolkit, we extracted four evolutionary features through mining information from PSSM in different aspects, including PSSM-composition, DPC-PSSM, PSSM-AC and RPSSM.
3. Model construction
To deal with the imbalanced classification problem for each of the features, we constructed 10 subsets by combining the positive samples and the same numbers of randomly selected negative samples from the training datasets. We accordingly trained 10 classifiers using the supported vector machine (SVM) and integrated them by averaging their prediction outputs. SVM is widely used to solve binary classification problems in the field of computational biology. Particularly SVM with a Gaussian radial basis kernel (RBF) has been successfully used for nonlinear biological sequence classification. Two parameters affect the performance of the RBF kernel SVM. Among them, Cost controls the cost of misclassification of data training, and Gamma is a specific parameter of the RBF kernel. In this study, for each SVM based classifier, the parameters Cost and Gamma were optimized using a grid search within the space {2-10,...,210}. In this way, we obtained an ensemble model as the baseline model for each feature (termed single feature-based model). To make full use of different types of evolutionary features, we averaged the prediction scores of their single feature-based models to form the final ensemble model.
4. Performance evaluation
The proposed method was rigorously and extensively validated based on the 5-fold cross-validation test, an additional independent test, and prediction capability was further investigated using case studies. Performance measurements include Sensitivity (SN), Specificity (SP), Accuracy (ACC), F-value, MCC, and the Receiver operating characteristic (ROC) curve as well as its AUC (Area under the curve) value. For a predictor, SN and SP measure its power of identifying positive and negative samples, respectively. ACC, F-value, MCC and AUC measure its comprehensive capability of identifying both positive and negative samples.
5. Server construction
The architecture of the PaCRISPR server consists of two major components: a client web interface and a server backend. The client web interface is responsible for interacting with users through the input and output displays, and to process the service logic including the illegal character detection, sequence validation and format. The former was implemented by JSP, CSS, jQuery, Bootstrap and their extension packages. Specifically, the sequence similarity was visualized by BlasterJS, and the phylogenetic tree was presented using jsPhyloSVG.The latter was implemented by the JAVA server development suite, including Struts 2 and Hibernate. The server backend is responsible for executing the whole prediction process, including encoding features, making predictions, and generating visualize-ready data. The feature encoding was implemented by using the DIFFUSER and POSSUM toolkits. The prediction program was written in R language dependent on the e1071 package for SVM modelling. The BLAST (version 2.8.1+) program was used to search against the known anti-CRISPRs for each predicted anti-CRISPR, and to record regions of their similarities for sequence similarity visualization. The MAFFT toolkit was used to generate multiple alignment results between each predicted anti-CRISPR and the known anti-CRISPRs for phylogenetic tree visualization. A Perl CGI program was written to string together these steps within a single thread. The client web interface interacts with the server backend through a fast and lightweight queueing system, implemented using the Gearman framework. The client web interface simply puts the user's submissions (each of them as a job) into the queueing system, where the Perl idle threads, maintained in a daemon thread pool with customizable size, pull and execute the jobs. During the whole process, the MySQL database is used to store the immediate and the final results, as well as synchronize messages between the client web interface and the server backend. In this way, the architecture brings better user experience by decoupling the client web interface that requires prompt response speed and the server backend that handles time-consuming jobs. This also makes the architecture amenable for expansions to add new computational facilities to meet the increasing demand in predicting ever accumulating genome-scale data.
ONLINE WEB SERVER
1. Using the PaCRISPR web server
PaCRISPR allows users to copy-and-paste or upload their interested sequences in FASTA format in the input page (shown in the following figure). When predicting query proteins, the PaCRISPR server provides two options in the Usage bar for different purposes. For normal use, the PaCRISPR server applies a built-in list of experimentally validated anti-CRISPRs to filter out the query proteins prior to using its models to execute the computational prediction. But for users who want to benchmark and test the prediction performance of the PaCRISPR server, they can disable the built-in list by selecting the For benchmarking test option to retrieve the prediction scores for all query protein sequences.
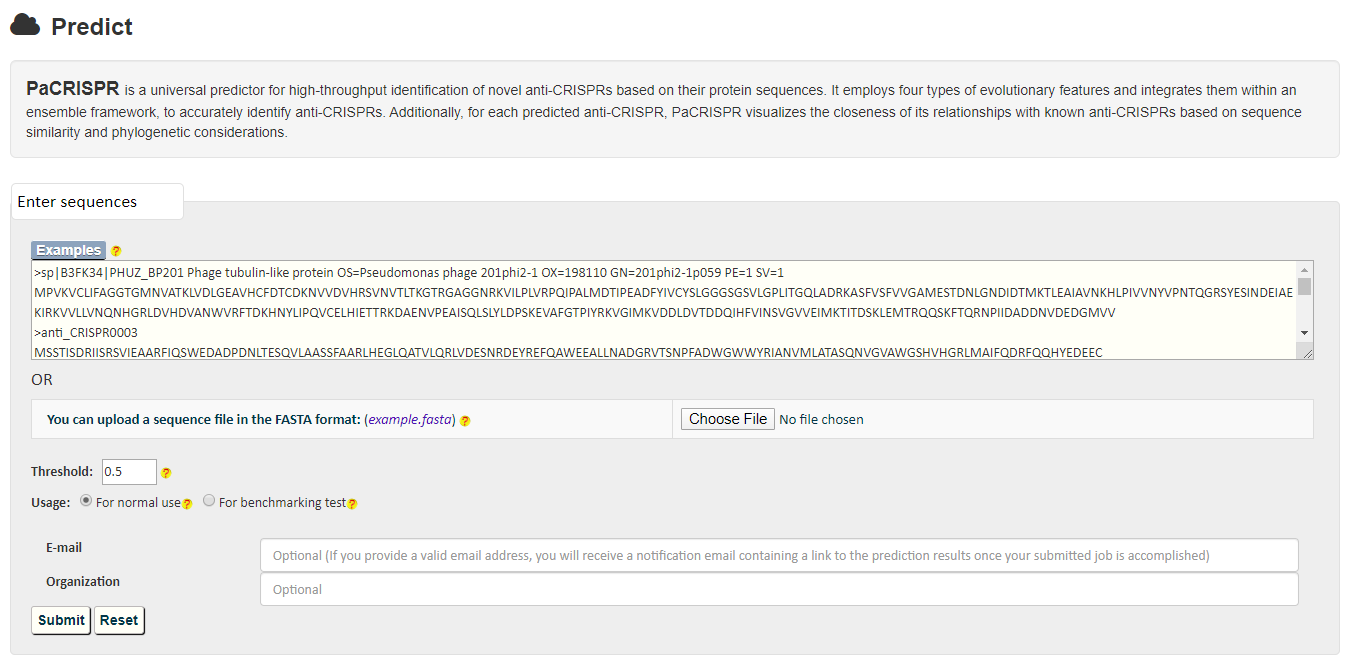
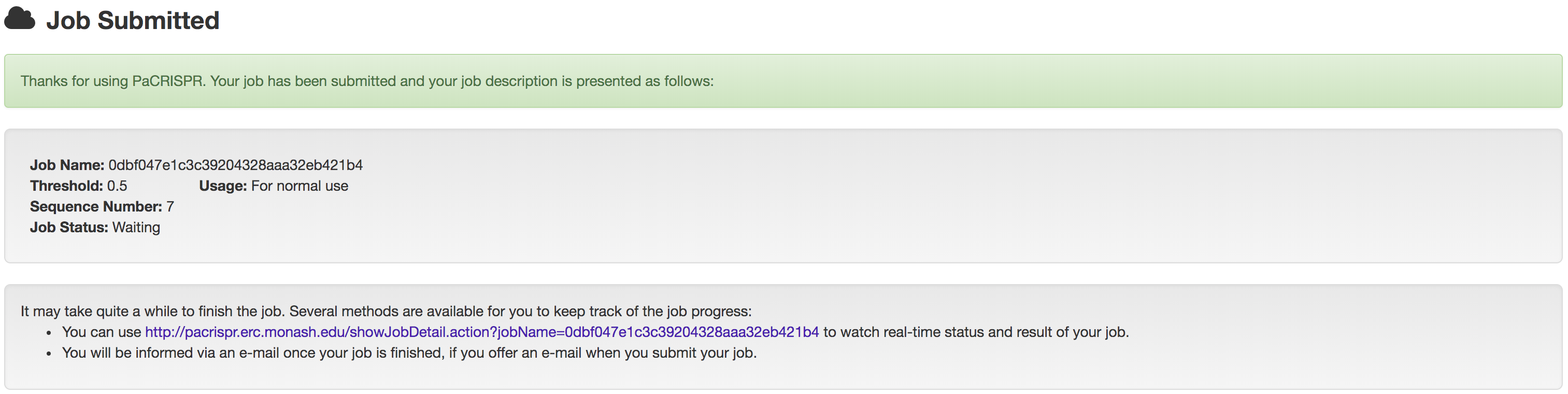
Once submitted, a unique link will be generated to refer to the job summary page during the job execution process (shown in the following figure). Users could use this link to track their job execution progress, and access or download their prediction results once completed.
1.1 Input Formats
Two types of input are accepted by PaCRISPR: sequences in FASTA format (strongly recommended) and raw sequences.
In the case of input sequences in the FASTA format, you can prepare and input them as follows:
MKLIVEVEETNYKNLVNYTKLTNESHNILVNRLISEYITKPYELRLDLSERYSNRDLIEFKFMLIEYCKEALQDIKELANSDEAYETDEAFEAVFRQLFEEVISNPDTVLKAFHSYTSFLEENK >AcrIII-1 III-A III-B Sulfolobus islandicus Athukoralage, 2020, Nature
MNKVYLANAFSINMLTKFPTKVVIDKIDRLEFCENIDNEDIINSIGHDSTIQLINSLCGTTFQKNRVEIKLEKEDKLYVVQISQRLEEGKILTLEEILKLYESGKVQFFEIIVD
In addition, the following input sequence, which is in the original format downloadable from the UniProt database:
VAAVKGTDGLTVDDYDQLSLDDLLELETFGLLFFVGSLE >AcrIIA11 II-A Clostridium sp. from human gut metagenome Forsberg, 2019, eLife MADMTLRQFCERYRKGDFLAKDRETQIEAGWYDWFCDDKALAGRLAKIWGILKGITSDYILDNYRVWFKNNCPMVGP
LYDDVRFEPLDEEQRDELYFGVAIDDKRREKKYVIFTARNDYENECGFNNVREVRQFINGWEDELKNEEFYKAREKKRQEMEEANNKFAEIMQRADEILWNLKED >AcrIIA13 II-A Staphylococcus schleiferi Watters, 2020, PNAS MEVMNKSIEIKDQNNIVLIDSLGQFFTDIENDNNGRYNIDYVLLNEVEHDNGNTYYEVGMYRTEEVPFSDKVTQDNVELLEDKWLQIDQQGESYV
ESIFFENEEDAREYIKLVLKGHETFEETAKAIGVIK
will be formatted (in order to remove those line breaks within the sequence) as follows:
In the case of raw sequences, you can input them as follows:
which will be formated by PaCRISPR as follows:
1.2 Input sequence limits
- The length of each submitted sequence should be in the range of 30 and 5000.
2. PaCRISPR Prediction Result Instructions
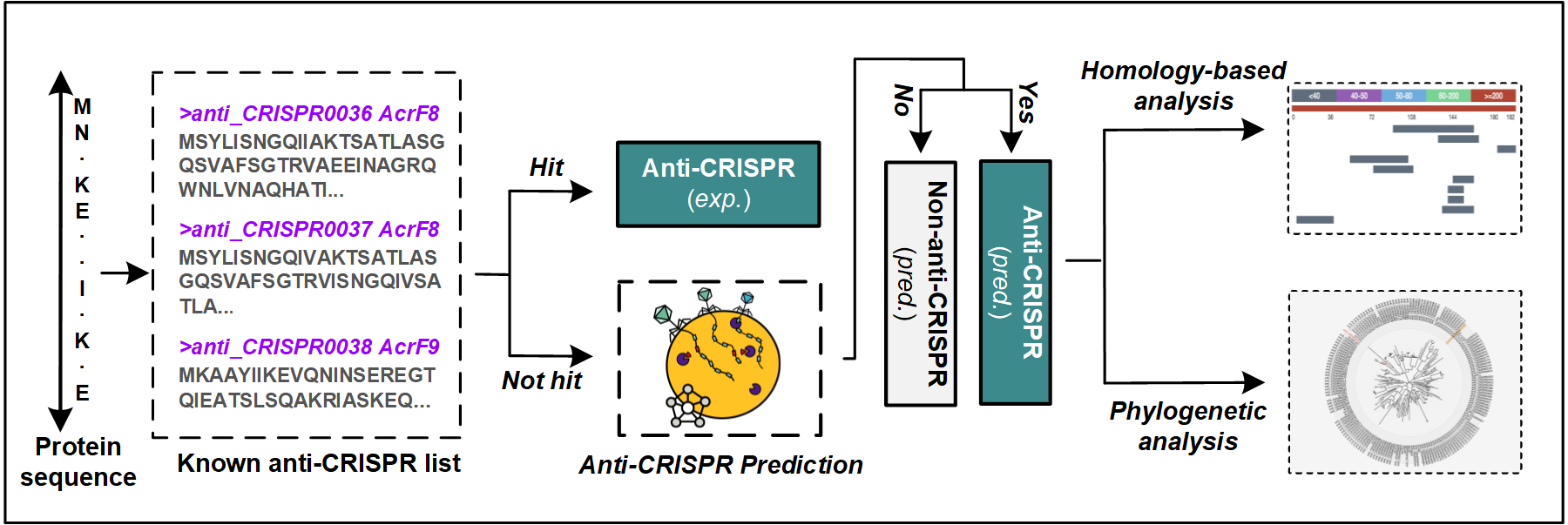
PaCRISPR contains a built-in list (continuously updated to keep in pace with new experimentally validated anti-CRISPRs) of anti-CRISPRs, as to annotate the prediction results after jobs are processed, through which we aim to distinguish the known anti-CRISPRs from the computationally predicted ones.
To distinguish between predicted and validated anti-CRISPRs, each of the proteins will be marked with either an Exp. if it has been experimentally validated or Pred. with a prediction score (an example is provided in the following figure).
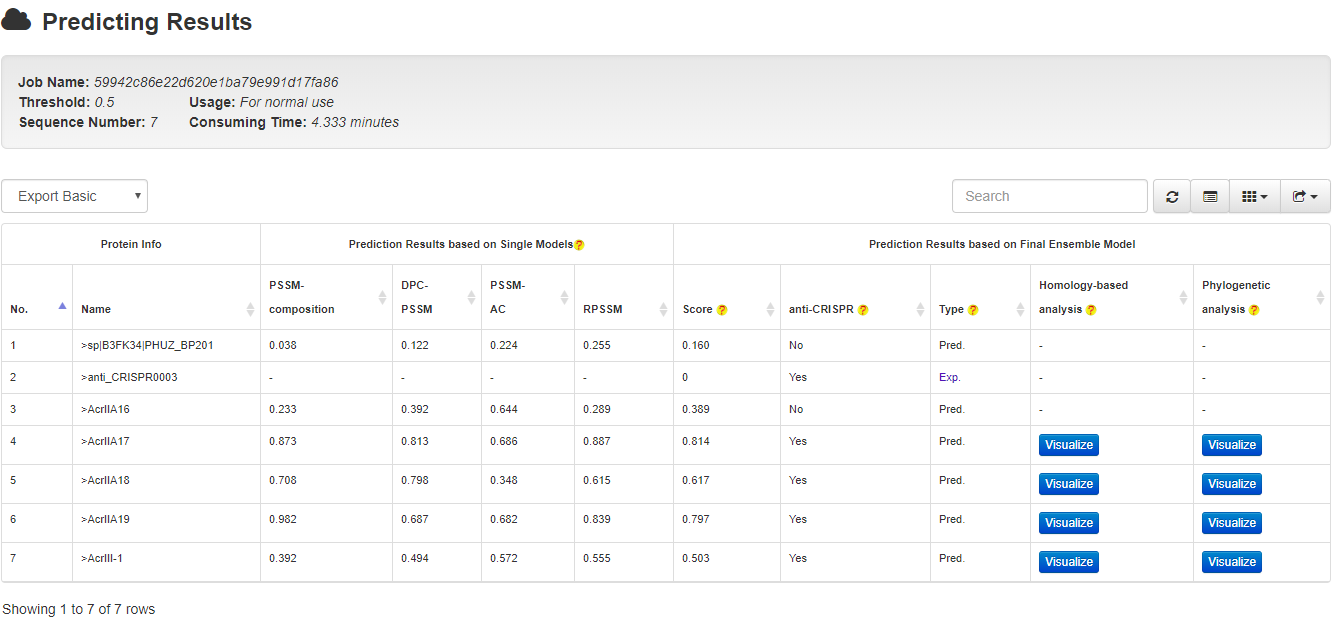
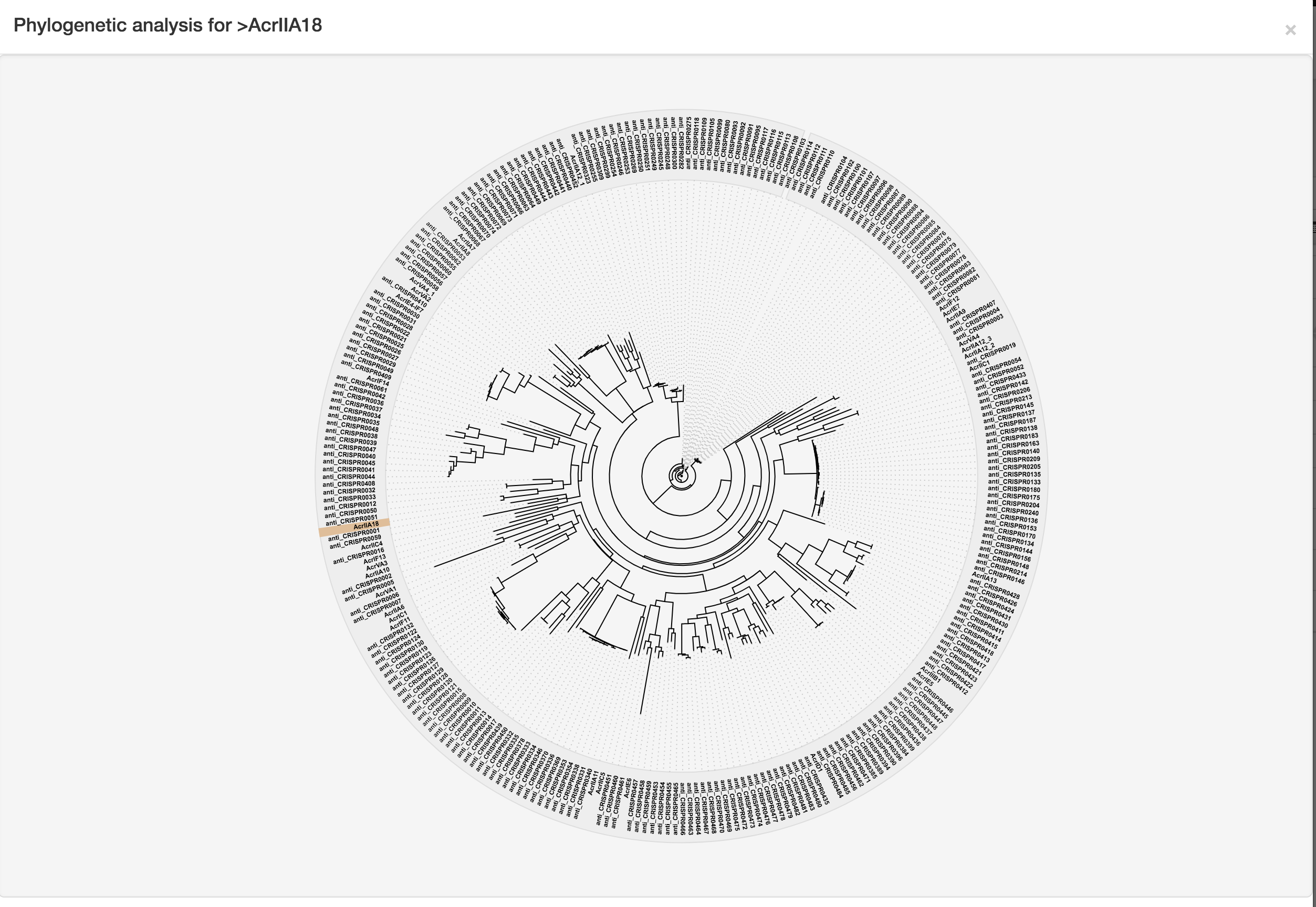
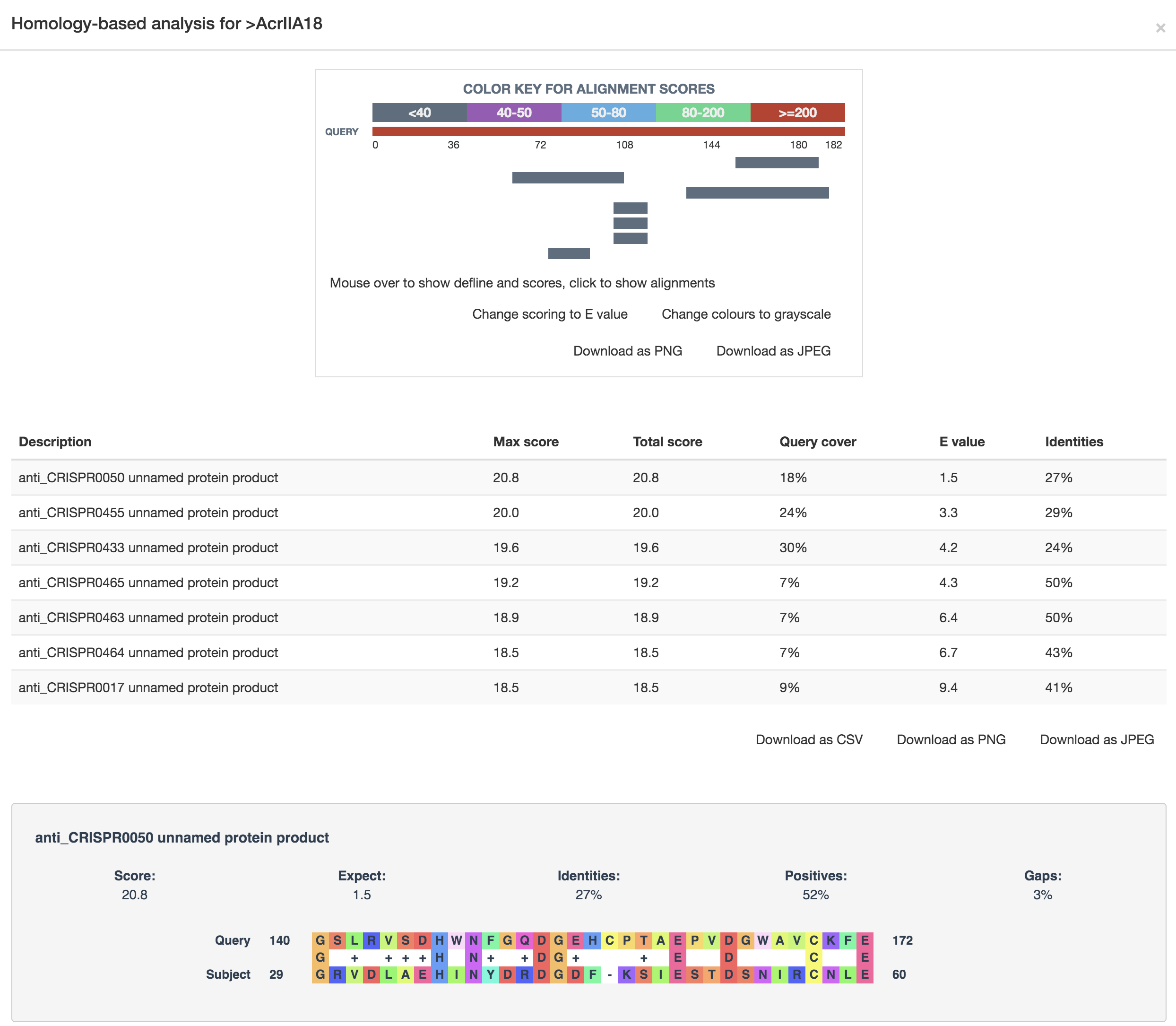
For validated anti-CRISPRs, users will be provided with a link to the relevant page in either the Anti-CRISPRdb or the unified online anti-CRISPR resource. To facilitate downstream analysis, for each predicted anti-CRISPR, we list the most homologous known anti-CRISPRs, chart their alignments, and highlights the pair-wise alignment. The interactive phylogenetic tree can instead illustrate its relationships to known anti-CRISPRs with the predicted anti-CRISPR highlighted. Each of the known anti-CRISPRs among the tree is marked with an external link to the Anti-CRISPRdb or the unified online anti-CRISPR resource, through which more detailed information could be obtained.
The sequence similarity relationship analysis was conducted by the blast-2.8.1+ program, followed by the visualization using the BLASTERJS library (an example is provided in the following figure).
The phylogenetic relationship analysis was conducted by the multiple sequence alignment program MAFFT, followed by the visualization using the jsPhyloSVG library (an example is provided in the following figure).